

Charting a Path to Accelerating Companion Diagnostic and Drug Development
Companion Diagnostics (CDx) are integral tools to enable precision medicine strategies. They allow clinicians to make the best treatment decision for a patient based on their unique biomarker profiles. Predictive biomarkers can be genetic mutations, protein expressions, or other molecular indicators associated with a particular disease that provide insights into a patient’s disease subtype, prognosis, and potential response to specific therapies. For instance, BRCA1/BRCA2 mutations predict the efficacy of PARP inhibitor treatment in specific breast cancer subsets. This personalized approach to patient selection not only enhances treatment effectiveness but also minimizes adverse effects, increases response rates, and reduces overall healthcare costs. Companion diagnostics are widely used in oncology, but their potential is expanding to other therapeutic areas as our understanding of disease mechanisms expands.
Despite the acknowledged benefits, translating biomarker-based CDx into clinical practice presents challenges. Fortunately, the innovative potential of CDx has united industry stakeholders, driving concerted efforts in development and approval to advance personalized treatment therapies for patients. Discovery Life Sciences continues to play an active role, supporting biomarker discovery and CDx development across the development continuum, from research through post-approval and beyond. Leveraging the world’s largest biospecimen network and multi-omics technologies, Discovery aids in identifying, characterizing, and validating candidate biomarkers and their corresponding CDx.
In a recent review article authored by scientists from Discovery’s Biomarker Services team titled ” Fast-tracking drug development with biomarkers and companion diagnostics,” McBrearty et al provide a comprehensive overview of key events in biomarker discovery and CDx development. The authors discuss strategies to streamline processes from early stages to clinical validation to fast-tracking CDx approval and their corresponding therapeutics. They cite key challenges and propose solutions for successful implementation of CDx to broaden their clinical utility, which will ultimately help improve patient outcomes.
Preclinical & Translational Testing
In large part, the regulatory approval of the CDx follows the contemporary drug approval process starting with a preclinical phase followed by clinical trials and regulatory approval (Figure 1).
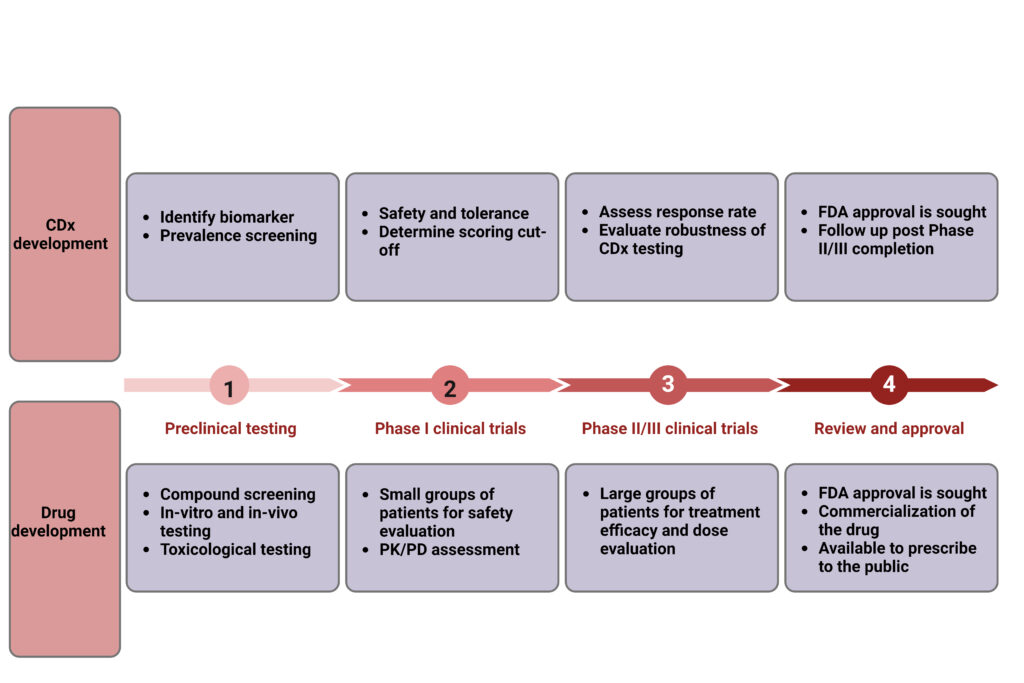
Figure 1. Typical course of CDx and drug development.
CDx development begins with biomarker discovery, utilizing various modalities such as immunohistochemistry (IHC), flow cytometry, and next-gen sequencing (NGS) to identify potential biomarkers associated with a specific disease. Once identified, these candidate biomarkers undergo validation using preclinical tumor models. However, some of these models lack the clinical relevance needed for successful translation of research data to clinical settings. Therefore, it is imperative that the preclinical tumor models used can accurately reflect the biological processes driving tumor growth in the intended patient population.
Researchers must adequately understand expression patterns to assess the prevalence of the CDx biomarker. Decisions made during early development should be based on data derived from high-quality study cohorts that are representative of the target population to ensure clinical relevance. Conducting large-scale sensitivity screenings using archived biospecimens or fresh patient tissue to assess biomarker expression levels in various cancer indications allows for prioritization of indications with the highest expression that are most likely to respond to the targeted therapy. Discovery’s access to their broad biospecimen network can expedite these evaluations during preclinical and translational assessments of CDx.
PHASE I: LOCKING IN THE CDx
During Phase I clinical trials, focus shifts towards assessing the safety and tolerance of the CDx. Researchers need to establish CDx scoring schemes and cutoffs that can be used to identify patients who are likely to respond to treatment with a drug. Developing an appropriate scoring scheme and cutoff for the diagnostic test during this phase is critical, as an ill-chosen scoring scheme and cut-off can significantly impact the outcome of a clinical trial.
In some cases, the presence or absence of certain biomarkers is sufficient to stratify patients into responders and non-responders, but for others like PD-L1 and Her2, the relative intensity of expression is important. Therefore, the CDx must be capable of discerning between negative, low/moderate, and high expression levels, as this distinction can dictate a patient’s eligibility for treatment (Figure 2).
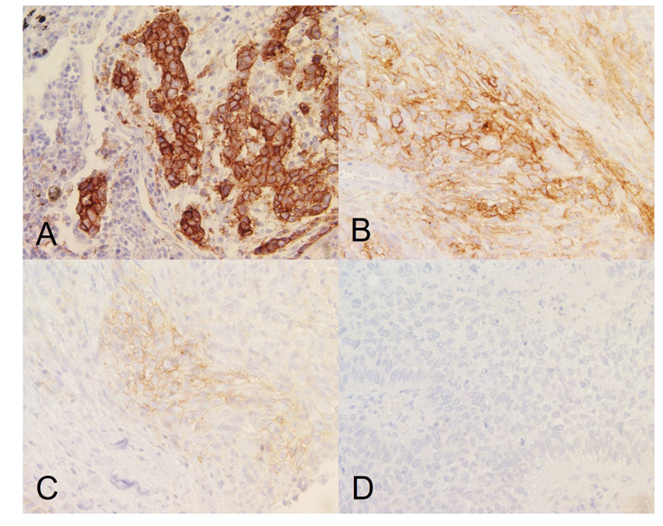
Figure 2. Tumor cell staining showing (A) high, (B) moderate, (C) low, and (D) negative PD-L1 membrane expression. Images taken at 40x magnification. Per FDA guidelines for Keytruda eligibility, tumors shown in (A-C) will be eligible for anti-PDL1 therapy.
The authors emphasize the importance of evaluating cutoffs and scoring schemes of the same CDx test used in different indications for inclusion/exclusion of patients. For instance, Her2 expression levels in gastric cancer differ from those in breast cancer due to variations in tumor biology, marker heterogeneity, and more common occurrences of incomplete membrane staining in gastric cancer. Different scoring systems are approved by regulatory agencies for breast and gastric indications to ensure accurate Her2-based patient selection.
For drugs targeting rare diseases, the CDx is often necessary as inclusion/exclusion criteria for clinical trial participation. Leveraging biomarkers to select patients likely to respond in N-of-1 trials can expedite the drug development process by allowing a single patient to be the sole point of analysis. While the FDA doesn’t always mandate simultaneous approval of a drug and its diagnostic, co-development is required when the CDx is used to select the patient population for clinical evaluation.
PHASE II/III CLINICAL TESTING: CONFIRMING CDX EFFICACY AND VERSATILITY
During Phase II and III clinical trials, CDx testing is evaluated for reproducibility and robustness in real-world settings. Reproducibility ensures consistent results across different laboratories and operators, while robustness ensures the CDx can reliably identify patient populations most likely to benefit from the drug and eliminate those unlikely to respond and suffer unnecessary side effects.
Discovery’s board-certified pathologists have a longstanding history of successful collaborations with biopharma clients in developing, validating, and implementing CDx assays for novel drug candidates. This includes establishing reproducible evaluation criteria and defining specific cut-off values crucial for determining patient eligibility in clinical trials and evaluating clinical outcomes like response rate or survival. Our approach has proven successful for leading CDx programs including Her2 and PD-L1.
CDx APPROVAL
Upon successful completion of clinical trials, CDx undergo regulatory approval from the FDA, followed by post-approval activities, which may include pathologist training, particularly for new biomarkers, and post-approval marketing. Our Pathology and Biomarker Academy provides peri- and post-approval services to help standardize assay interpretation, train and educate pathologists and histotechnicians, and help increase adoption post-market. Because many CDx target the same biomarker, having a CDx approved for one drug means that testing is more streamlined for the CDx for similar classes of drugs. The approval of Osimertinib, an EGFR tyrosine kinase inhibitor (TKI), is the most notable example of leveraging a previously approved CDx to accelerate the approval process of a new drug. Compared to the average approval time of 10-15 years from discovery to drug approval, the timeline for Osimertinib was condensed to just six years.
While tissue-based biopsy tests currently dominate approved CDx, liquid biopsy tests analyzing circulating tumor DNA (ctDNA) are emerging as important tools in precision medicine. However, discordance in ctDNA tests compared to traditional tissue biopsies, including a higher rate of false negatives, has thus far presented setbacks to establishing their clinical utility. Therefore, matched FFPE samples from the same patient serve as a critical benchmark for the validation and qualification during ctDNA companion diagnostic development. Discovery now offers double-spun plasma, FFPE, and urine matched sets across numerous oncology indications. Through our global Discovery Partners® network, we can procure and process prospective collections of matched biospecimens with custom criteria to support your liquid biopsy projects. Read more about Discovery’s liquid biopsy capabilities in our new white paper.
The journey of companion diagnostics (CDx) from discovery to approval is a critical component in advancing personalized medicine and expediting the development of innovative therapies. With Discovery’s expertise in biomarker discovery, assay development, and clinical validation, we are poised to support your CDx project from bench to bedside. Contact a Discovery CDx specialist today to kickstart your project.